
DSB repair around the clock
DSBs arise constantly in our cells by endogenous cellular mechanisms or are induced by exogenous agents, including ionising radiation (IR) and chemotherapeutics. Cells react to these lesions by inducing cell cycle arrest and initiating DSB repair mechanisms. The faithful repair of these breaks is critical for cell survival and genomic integrity. Incorrect repair leads to the accumulation of mutations and the development of diseases such as cancer. Furthermore, incomplete or inactive repair leads to the persistence of unrepaired breaks and subsequent induction of cell death, a process often exploited for cancer therapy. Therefore, elucidating DSB repair mechanisms is central to understanding the pathogenesis of diseases and evaluating and improving the efficacy of treatment. Since breaks are channelled into different repair mechanisms depending on the cell-cycle phase, we employ cell cycle-specific analysis of DSB repair to gain a more accurate knowledge of these processes. We use various cell and molecular biology techniques to study repair factors and their role in response to IR-, chemotherapeutics- and enzyme-induced DSBs in multiple cell systems.
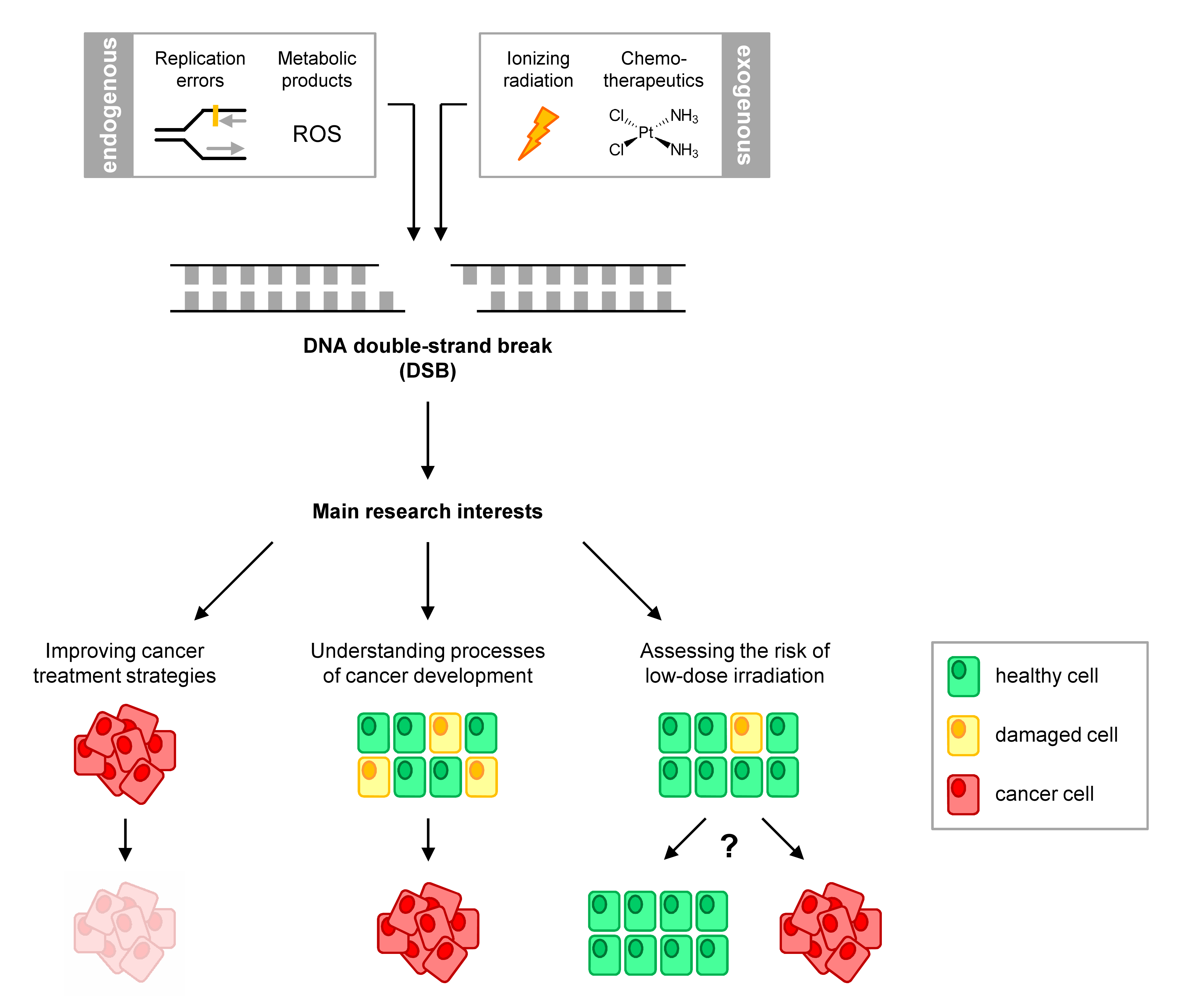
Targeting homologous recombination for cancer therapy
Homologous recombination (HR) is an error-free DSB repair pathway that is active in the S/G2 phase of the cell cycle and is the main pathway repairing spontaneously arising breaks. HR is often inactivated in cancer, a feature that has been central to various therapeutic strategies. Our lab has identified various novel factors involved in HR regulation, function and HR sub-pathway choice. We also characterized the usage of distinct HR sub-pathways in specific types of cancer compared to normal cells. Current projects involve the advancement of these findings to in vivo mouse models and to the pre-clinical evaluation of targeting these factors. We are particularly interested in identifying novel synthetic lethality relationships that could open up opportunities for therapeutic intervention.
Whole-genome mapping of NHEJ mechanisms and fidelity
Non-homologous end-joining (NHEJ) is a main DSB repair pathway operating throughout the cell cycle and is known to be error-prone. The compromised fidelity of this pathway is believed to cause mutations that likely drive cancer initiating events. Our lab identified a sub-pathway of NHEJ that shares features with HR and harbours potential for error-free repair. We are currently investigating factors affecting choice of NHEJ sub-pathways as well as the mechanisms driving enhanced repair accuracy. Current projects employ next-generation sequencing (NGS) for the genome-wide mapping of mutational signatures of NHEJ to identify mutation rates in wild-type and NHEJ-mutant cells. We also use chromatin immunoprecipitation sequencing (ChIP-Seq) to elucidate repair mechanisms operating at specific genomic loci and the influence of the chromatin landscape on repair pathway choice.
Evaluation of cellular responses following low-dose irradiation
While we encounter high doses of IR in specific situations, such as radiotherapy, we are constantly exposed to low IR doses in our daily lives from sources such as background radiation (e.g. radon) and routine medical examinations (e.g. X-ray diagnostics). Our work revealed that cells do not repair lesions from such exposure efficiently and possibly use distinct mechanisms from those handling higher levels of damage. This discrepancy could underlie an enhanced risk to low-dose IR exposure, in the form of increased mutations or, alternatively, can represent a protective mechanism where damaged cells are eliminated by cell death. Therefore, it is important to study the exact molecular processes involved in these responses in order to improve IR risk assessment and radiation protection procedures.
Funding and Collaborations


